Serotonin Synthesis Amplification
Serotonin
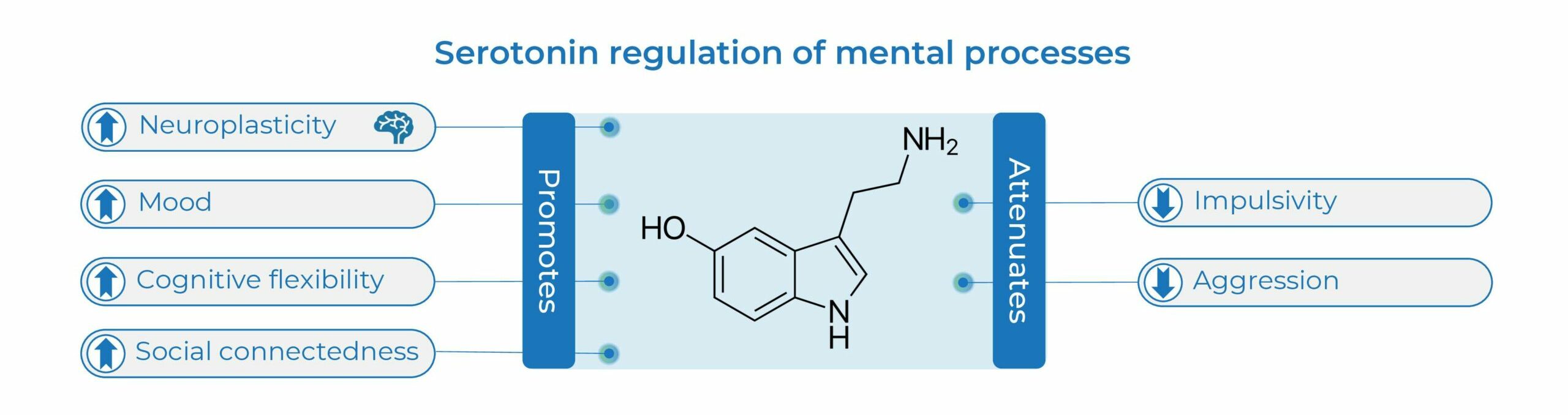
The neurotransmitter serotonin plays multiple roles in the brain supporting mental health. Serotonin is a potent endogenous neuroplastogen, on the synaptic, neurotrophic, circuit, anatomical, and phenomenological levels (1, 2). Serotonin promotes mood, cognitive flexibility, and sociability (3-5), and attenuates aggression and impulsivity (6). Serotonin Synthesis Amplification aims to strengthen overall endogenous serotonin function.
Serotonin Synthesis Amplification
Serotonin Synthesis Amplification denotes a pharmacological treatment that amplifies brain serotonin synthesis, resulting in a higher concentration of serotonin neurotransmitter molecules in the brain, and overall amplified brain serotonin activity – in effect, creating a stronger, more resilient brain serotonin system.
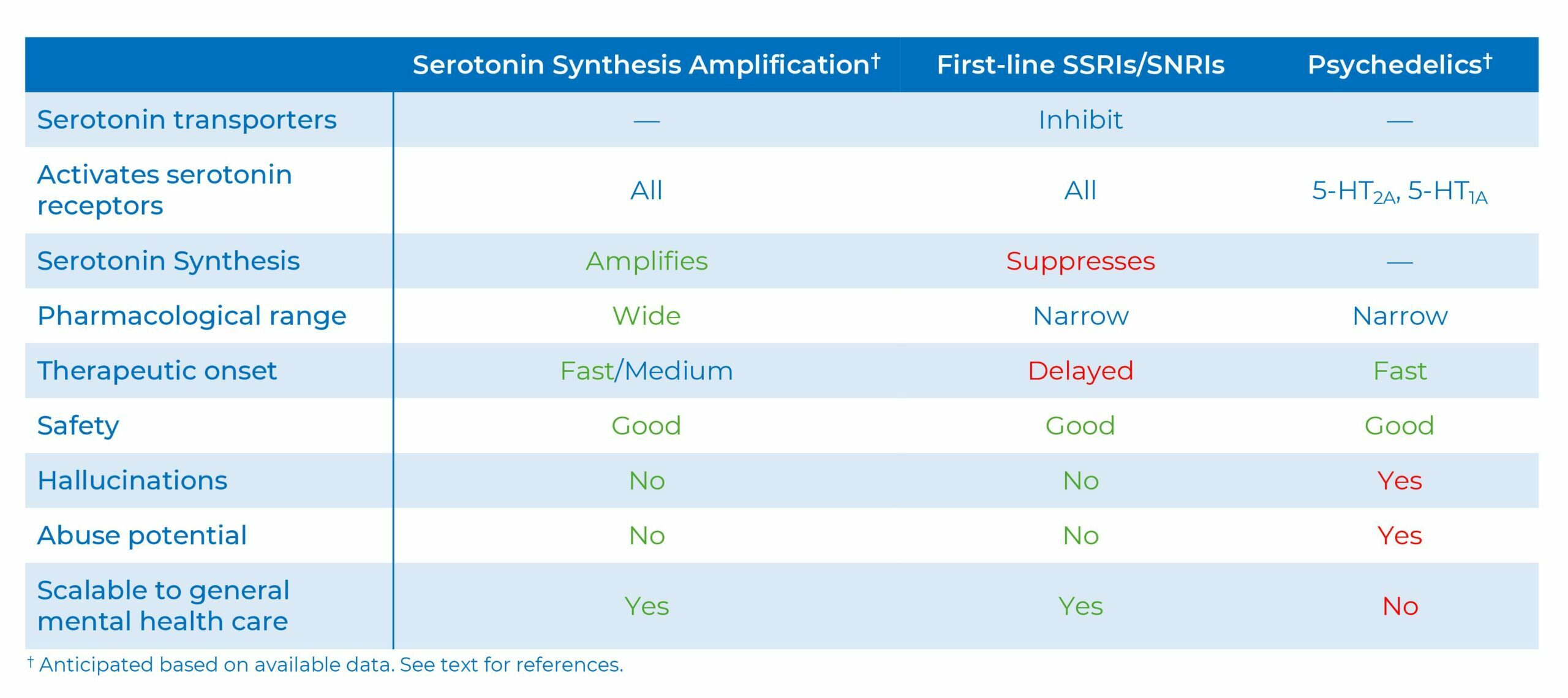
Serotonin Synthesis Amplification is distinct from existing serotonergic pharmacologies, e.g., selective serotonin reuptake inhibitors (SSRIs), which remove a “brake” on serotonin neurotransmission, and psychedelics, which activate select serotonin receptors.
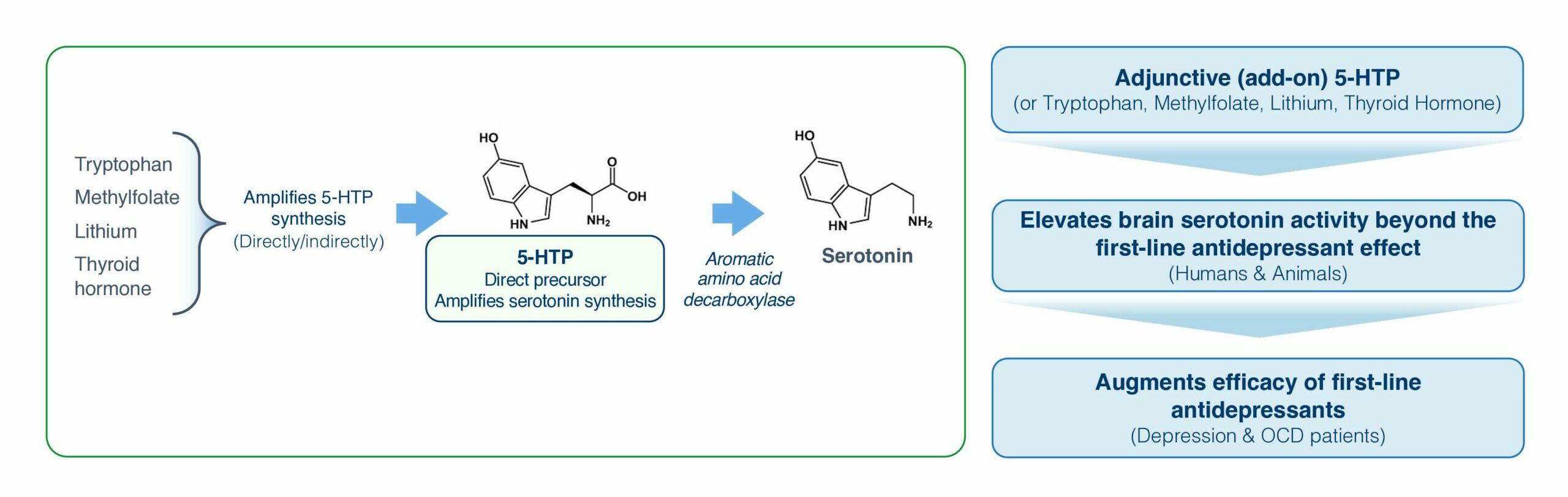
Serotonin Synthesis Amplification is reported to have therapeutic potential across a number of brain disorders (7-27). Notably, more than 30 published clinical studies report that compounds causing Serotonin Synthesis Amplification augment antidepressant efficacy in depression or obsessive-compulsive disorder (OCD) patients responding inadequately to first-line antidepressants, e.g., (10, 28-31). Serotonin Synthesis Amplification per se have a good 60 y human (8, 32, 33). Hence, Serotonin Synthesis Amplification represents a risk-mitigated antidepressant augmentation pharmacology. Indeed, Serotonin Synthesis Amplification has been pursued since the 1970s, using as the active compound either 5-hydroxytryptophan, 5 HTP (the natural rate-limiting serotonin precursor) (11), tryptophan (5-HTP precursor) (28), methyl-folate (amplifies 5-HTP synthesis) (29), lithium (amplifies 5-HTP synthesis) (34), or thyroid hormone (amplifies 5-HTP synthesis) (35). However, all five compounds have limitations precluding general clinical implementation and regulatory approval as adjunctive antidepressants.
Evecxia is the first company realizing the therapeutic potential of Serotonin Synthesis Amplification
Serotonin Synthesis Amplification is supported by dozens of clinical studies for safety and efficacy, particularly as adjunctive antidepressant therapy. Evecxia is deploying pharmaceutical approaches to enable sufficient and sustained 5-HTP plasma exposure, enabling safe, sufficient, and sustained Serotonin Synthesis Amplification, thereby realizing its therapeutic potential. Among the compounds known to cause Serotonin Synthesis Amplification, 5 HTP is clearly advantaged by being the direct serotonin precursor and have no off-target pharmacology. But native 5-HTP is a poor drug due to minimal oral bioavailability and rapid elimination from the circulation.
Evecxia’s lead drug candidate EVX-101 enables sufficient and sustained 5-HTP exposure at twice daily oral dosing, to treat patients with depression or OCD responding inadequately to first-line antidepressants. Second drug candidate EVX-301 enables fast-onset sustained 5-HTP exposure via intravenous (I.V.) dosing, to treat patients hospitalized with suicidal ideation crises.
Serotonin Synthesis Amplification is distinct from first-line serotonin reuptake inhibitor antidepressants
First-line selective serotonin reuptake inhibitor (SSRI) and serotonin-norepinephrine reuptake inhibitors (SNRI) antidepressants work predominantly by blocking the serotonin transporter (SERT). SERT is a natural “brake” on serotonin activity. However, in contrast to rodents, human forebrain SERT levels are very low (36, 37), suggesting SERT is not a major serotonin regulator in the human brain. Indeed, in humans, SSRIs only elevate serotonin release (and hence activity) moderately, inconsistently, and with a delay (38-40) - which could, in part, explain why SSRIs (and SNRIs) treat depression, OCD, and other mood disorders modestly, inconsistently, and with a delay (41-43). Further, SSRIs/SNRIs have a narrow dynamic range, i.e., even at low label doses SSRIs/SNRIs approach saturation occupancy of the SERT (44), consistent with that SSRI or SNRI dose-increases yield limited added antidepressant efficacy. Moreover, rodent studies suggest long-term SSRIs at therapeutically relevant doses substantially suppress brain serotonin synthesis and levels (45), which, if occurring in patients, could constrain clinical efficacy. In contrast, Serotonin Synthesis Amplification amplifies serotonin levels and neurotransmission, over a wide dynamic range, with fast-to-medium onset.
Serotonin Synthesis Amplification is distinct from psychedelics
Psychedelics activates specific serotonin receptors, notably 5-HT2a receptors, which is associated with hallucinations, abuse potential, and apparent fast-onset antidepressant efficacy when combined with therapy; although, unblinding and expectation-bias could contribute to the antidepressant effect (46-48). In contrast, Serotonin Synthesis Amplification activates the full complement of serotonin receptors and appears devoid of hallucinations and abuse potential.
Serotonin Synthesis Amplification augments first-line antidepressant effect in dozens of clinical trials
Serotonin Synthesis Amplification is arguably the best clinically validated antidepressant augmentation pharmacology not used in any approved or late development stage antidepressant. When added to ongoing first-line serotonin reuptake inhibitor therapy, compounds inducing Serotonin Synthesis Amplification elevate serotonin activity (i.e., extracellular serotonin levels) beyond the first-line effect, including 5-HTP (49, 50), tryptophan, methyl-folate (51), lithium (52), and thyroid hormone (35). At least 30 published clinical trials report adjunctive therapy with any of these compounds can augment antidepressant efficacy when first-line serotonin reuptake inhibitors alone are insufficient, e.g., (10, 28-30, 53). These clinical findings are promising. More so since no adequately powered antidepressant augmentation trials using optimized doses, dosage forms, and dosing regimens have been executed for any of these compounds, which are required to establish the possible therapeutic effect size of a novel pharmacology.
Mechanism and short-comings of previous approaches to Serotonin Synthesis Amplification
To elevate serotonin activity beyond the first-line serotonin reuptake inhibitor antidepressant effect, to treat depression patients responding inadequately, has been pursued from the 1970s (11, 28) till the present (29, 54), by adjunctive (add-on) administration of a compound inducing Serotonin Synthesis Amplification:
- 5-hydroxytryptophan (5-HTP) is advantaged by being the direct, rate-limiting precursor to serotonin (7). 5-HTP powerfully and over a wide dynamic range induce Serotonin Synthesis Amplification (49, 55). 5-HTP is specific to serotonin, with minimal off-target effects on other neurotransmitter systems (49, 55). Further, 5-HTP has a remarkable good 60-year human safety record (7, 32). The native 5-HTP molecule is disadvantaged by poor drug properties, i.e., low oral bioavailability (~5%) and rapid elimination (T1/2 ~3h) (56), likely explaining why no 5-HTP drug product has ever been developed or FDA-approved.
- Tryptophan is the precursor of 5-HTP; but, only a few percent of tryptophan is metabolized to 5-HTP. Tryptophan is modestly effective in inducing Serotonin Synthesis Amplification. Further, high-dose tryptophan (>5 gram/day), as needed to amplify serotonin, could impact multiple metabolic and neurotransmitter systems (57).
- Methyl-folate is a co-factor in 5-HTP synthesis (51). But co-factor administration is unlikely to potently amplify serotonin and may mainly treat depression patients a priori deficient in folate (58, 59).
- Lithium amplifies brain serotonin synthesis, as reported repeatedly since the 1970s (60, 61). There is consensus that amplified serotonin activity is critical for adjunctive lithium’s well-established antidepressant augmentation efficacy (34). Indeed, adjunctive lithium to serotonin reuptake inhibitors augments brain serotonin release, and hence serotonin activity, beyond the SSRI-alone effect in rodents (52). But lithium has multiple biological effects and is not approved for depression. Lithium is sparingly used in depression due to safety concerns and a requirement for tight monitoring (62).
- Thyroid hormone facilitates serotonin activity (35). Hypothyroidism may lower while thyroid hormone administration may increase brain serotonin levels (63, 64). While data is less plentiful than for lithium, there is consensus that amplified serotonin activity is critical for adjunctive thyroid hormone’s well-established antidepressant augmentation efficacy (35). Indeed, adjunctive thyroid to serotonin reuptake inhibitor antidepressants augment brain serotonin release, and hence serotonin activity, beyond the antidepressant-alone effect in rodents (65). But thyroid hormone has multiple biological effects and is not approved for depression. Thyroid hormone is sparingly used in depression due to safety concerns and a requirement for tight monitoring (66).
Realizing Serotonin Synthesis Amplification Therapy By Realizing The Therapeutic Potential Of 5-HTP
The optimal active compound to use in pharmaceutical technologies designed to produce Serotonin Synthesis Amplification is 5-hydroxytryptophan, 5-HTP. 5-HTP (i) is the direct rate-limiting precursor of serotonin, (ii) potently amplifies serotonin synthesis, (iii) has minimal off-target, and (iv) has a favorable human safety record (7, 32, 49, 55). But 5-HTP has low oral bioavailability and undergoes rapid elimination (56), making the naked 5-HTP molecule impractical as a therapy. To enable practical Serotonin Synthesis Amplification therapy, sufficient and sustained 5-HTP plasma exposure is required, to enable sufficient and sustained Serotonin Synthesis Amplification.
Evecxia is deploying proprietary pharmaceutical technologies to ensure sufficient and sustained 5 HTP plasma exposure to enable sufficient and sustained Serotonin Synthesis Amplification. EVX-101 is an oral tablet, comprising (i) 5-HTP as the active compound, (ii) low-dose carbidopa as a 5-HTP absorption-multiplier, (iii) gastro-retention to enable (iv) sustained-release delivery of 5-HTP/carbidopa to the upper intestine, the intestinal region where 5-HTP is absorbed. EVX-101 is in development for adjunctive Serotonin Synthesis Amplification therapy in depression and obsessive-compulsive disorder (OCD) therapy when first-line serotonin reuptake inhibitor antidepressants alone are inadequate. EVX-301 is an intravenous infusion of 5-HTP that ensures sufficient and sustained 5-HTP plasma exposure with fast onset. EVX-301 is in development for suicidal ideation crisis.
5-HTP is the natural rate-limiting precursor of serotonin
5-HTP is synthesized in the body from tryptophan derived from the diet. 5-HTP synthesis is mediated by tryptophan hydroxylase (using methyl-folate as a co-factor) and is the rate-limiting step in serotonin synthesis. In the body 5-HTP is rapidly converted to serotonin by the high-capacity promiscuous enzyme aromatic amino acid decarboxylase (7). Circulating 5-HTP levels are usually extremely low (56).
5-HTP administration amplifies brain serotonin synthesis
Numerous animal and human studies demonstrate that 5-HTP administered orally or parenterally reaches the brain, via active transport, and is converted to serotonin by serotonergic neurons, thereby amplifying serotonin synthesis, levels, and neurotransmission, e.g., (49, 50, 55, 67-69).
5-HTP has shown therapeutic promise in mood disorders
It is well-recognized that 5-HTP has shown therapeutic promise, particularly in depression (7-9). Importantly, several blinded and open-label studies report that adjunctive 5 HTP augments antidepressant efficacy when first-line serotonin reuptake inhibitor antidepressants alone are inadequate (10-16). 5-HTP has further been reported to treat anxiety (17) and obsessive-compulsive disorder (OCD) (18).
These therapeutic findings with 5-HTP are promising; not least since (i) 5-HTP as the native molecule is a poor drug, due to poor absorption and rapid elimination, and (ii) appropriate dosage forms, dosages, and dosing regimens—mandatory to establish therapeutic potential—has never been established for 5-HTP. Plausibly, the previous studies of 5-HTP in mood disorders have not revealed the full therapeutic potential of Serotonin Synthesis Amplification therapy using 5-HTP as the active compound.
5-HTP has shown therapeutic promise in other CNS disorders
5-HTP therapy has been used extensively off-label to treat myoclonus (19, 20). 5-HTP therapy has also been reported to treat obesity (21), fibromyalgia (22), migraine (23), sleep disorders (24), and certain childhood developmental disorders (25, 26). With the FDA’s consent, 5-HTP is currently being used in the U.S. off-label to treat rare inborn errors of the metabolism of serotonin and other monoamines (27).
5-HTP has a favorable human safety record
5-HTP has a remarkably good human safety record. 5-HTP has been administered to thousands of human subjects in hundreds of studies since the 1950s (PubMed). Many studies administered high (>1 gram/day) 5-HTP doses, often for long durations (months, years), and often with other drugs, including serotonin reuptake inhibitors and other antidepressants. Yet, 5 HTP has not been reported to cause any severe side effects, human disease, or deaths (7, 8, 32).
In the 1990s, tryptophan and 5-HTP were briefly suspected to cause eosinophilia-myalgia syndrome (EMS). Subsequently, the cases of EMS were conclusively determined to be caused by an impurity (“peak X”) in lots from a specific supplier, not by tryptophan or 5-HTP. No EMS cases have since been associated with tryptophan and 5-HTP (32). Two case reports associated high doses of 5-HTP and carbidopa (aromatic amino acid decarboxylase inhibitor) with scleroderma (70, 71); however, a temporal/causal relationship with 5-HTP was not established. Scleroderma is an auto-immune disease, can arise spontaneously, and has been associated with numerous different drug therapies (72). The clinical literature contains no reliable reports of serotonin syndrome associated with 5-HTP, suggesting 5-HTP have low propensity to cause this adverse event in humans.
Recently, Evecxia completed a Phase 1 SAD/MAD trial for EVX-101 and a Phase 1 SAD trial for EVX-301 both in healthy volunteers treated with a first-line antidepressant (escitalopram, an SSRI). Both trials reported excellent safety profiles.
5-HTP has a favorable non-clinical safety record
A rat 12-month toxicology study of high-dose oral 5-HTP (875 mg/kg/day) via the drinking water (which distributes 5-HTP administration over the day) found no toxicological effects on any organ class or parameter (73). Likewise, Evecxia’s 3-month GLP toxicology studies of EVX-101 in rats and pigs treated with an SSRI found no toxicological effects, despite sustained plasma levels of 5-HTP exceeding 2000 ng/ml, 20-fold higher than the putative human therapeutic exposure target of 100 ng/ml (56).
In rodents, high parenteral acute bolus doses of 5-HTP (e.g., 100 mg/kg) in combination with an SSRI can cause transient “serotonin syndrome” (74). However, this is an artifact of preclinical pharmacology methodology, i.e., extreme bolus doses and nonoral routes of administration. Similar high parenteral bolus doses of Prozac and caffeine kill rodents (75), but Prozac and coffee are safe in humans.
The native 5-HTP molecule has poor drug properties
Native 5-HTP is a poor drug. First, 5-HTP has low bioavailability, at ~5%. Second, 5-HTP is very short-acting, with an elimination half-life of ~3 hours (56). To ensure sufficient and sustained 5-HTP plasma exposure, four to six high daily doses of native 5-HTP would be required, which is infeasible in clinical practice. Co-administration of separate tablets of high-dose carbidopa multiplies 5-HTP’s bioavailability, but only moderately prolongs 5-HTP’s half-life, while causing rapid 5-HTP CMax-spikes associated with increased adverse events (76). Third, 5-HTP is only absorbed by the upper intestine, precluding using conventional (colon-dependent) sustained-release formulations to extend 5-HTP delivery and exposure (56). These limitations plausibly explain why no 5-HTP drugs was ever developed.
No FDA-approved 5-HTP drug exists
Botanical 5-HTP is available as a dietary supplement. Dietary supplements are not drugs. Dietary supplements are not FDA-approved for therapy and are not monitored, guaranteed for integrity, purity, drug delivery, safety, efficacy, and absence of adulterants by the FDA (77). Botanical 5-HTP is extracted from the seeds of Griffonia Simplicifolia harvested in the wild in West Africa, the future supply of which remains uncertain (78). Evecxia has established a proprietary 5-HTP chemical synthesis pathway and 5-HTP active pharmaceutical ingredient (API). EVX-101 and EVX-301 will be marketed using Evecxia’s proprietary 5-HTP API (56).
EVX-101 and EVX-301 would be first FDA-approved 5-HTP drug products
EVX-101 and EVX-301 remedy 5-HTP’s limitations as a drug, enabling sufficient and sustained 5 HTP plasma exposure, for the first time enabling sufficient and sustained Serotonin Synthesis Amplification therapy. EVX-101 and EVX-301 are being developed as drugs according to FDA guidance, would be approved for therapy by the FDA based controlled clinical trials, and would be manufactured and marketed as prescription (Rx) drugs under FDA supervision.
5-HTP

Serotonin

SSRI

Serotonin Transporter
Serotonin Receptor

Baseline
Serotonin is continuously released extracellularly into the synaptic cleft. Extracellular (active) serotonin activates post-synaptic receptors that mediates serotonin neurotransmission. Pre-synaptic serotonin transporters work as a break on serotonin neurotransmission, by pumping extracellular serotonin back into the pre-synaptic neuron terminal.
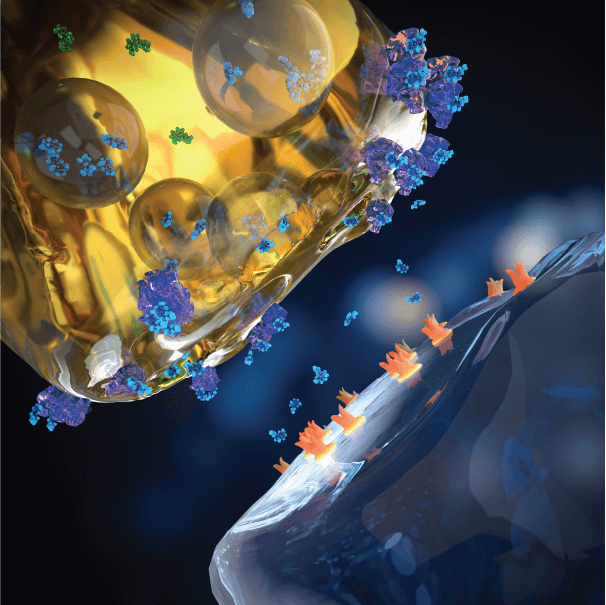
SSRI treated
Blocking the serotonin transporter with a serotonin reuptake inhibitor, SSRI (or another serotonin reuptake inhibitor) elevates extracellular (active) serotonin in the synapse. But serotonin transporter levels are low in the human forebrain and SSRIs only elevate extracellular serotonin (and serotonin activity) modestly, inconsistently, and with a delay, likely contributing to the modest, inconsistent, and delayed therapeutic response to SSRIs in patients.
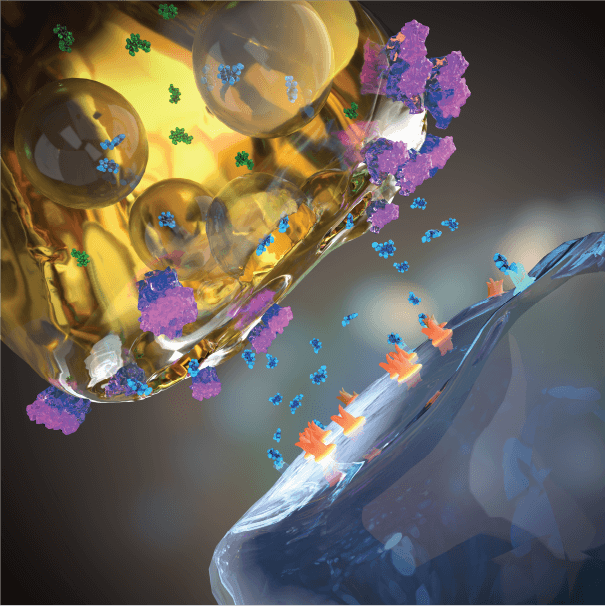
Adjunctive EVX-101/EVX-301 to SSRI treatment
Adjunctive EVX-101 and EVX-301 amplifies neuronal serotonin synthesis and release, thereby synergizing with SSRI (or SNRI) treatment in elevating extracellular (active) serotonin and amplifying serotonin neurotransmission potently beyond the SSRI effect, which 30 years of clinical data support augment antidepressant efficacy in mood disorder patients.
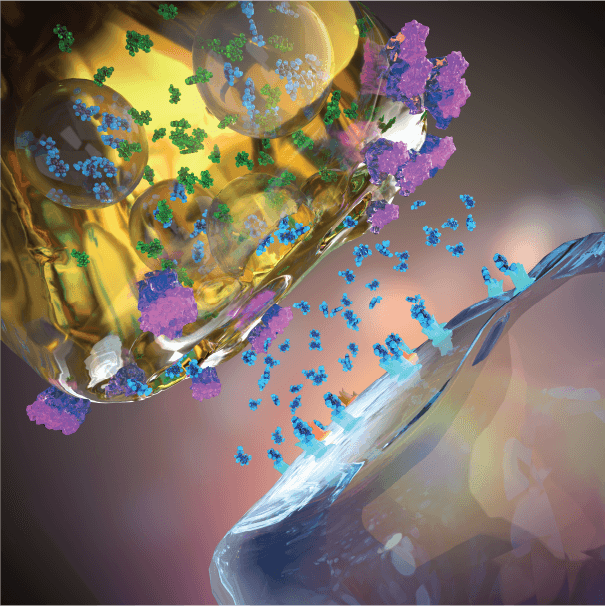
References & Abbreviations
-
-
- Kraus C, et al. (2017): Neuroscience and biobehavioral reviews. 77:317-326.
- Harmer CJ (2008): Neuropharmacology. 55:1023-1028.
- Puhringer W, et al. (1976): Neurosci Lett. 2:349-354.
- Clarke HF, et al. (2004): Science. 304:878-880.
- Duerler P, et al. (2022): J Neurochem. 162:60-79.
- Coccaro EF, et al. (2015): CNS Spectr. 20:295-302.
- Turner EH, et al. (2006): Pharmacol Ther. 109:325-338.
- Jacobsen JPR, et al. (2016): Trends Pharmacol Sci. 37:933-944.
- Shaw K, et al. (2002): Cochrane Database Syst Rev.CD003198.
- van Praag HM (1982): Adv Biochem Psychopharmacol. 34:259-286.
- van Praag HM, et al. (1974): Psychopharmacologia. 38:267-269.
- Nardini M, et al. (1983): Int J Clin Pharmacol Res. 3:239-250.
- van Hiele LJ (1980): Neuropsychobiology. 6:230-240.
- Moron P, et al. (1978): Psychologie medicale. 10:783-794.
- Zmilacher K, et al. (1988): Neuropsychobiology. 20:28-35.
- Kious BM, et al. (2017): J Clin Psychopharmacol. 37:578-583.
- Kahn RS, et al. (1987): Int Clin Psychopharmacol. 2:33-45.
- Yousefzadeh F, et al. (2020): Int Clin Psychopharmacol. 35:254-262.
- Levy A, et al. (2016): Curr Treat Options Neurol. 18:21.
- Magnussen I, et al. (1982): Acta Neurol Scand. 66:276-282.
- Ceci F, et al. (1989): J Neural Transm. 76:109-117.
- Caruso I, et al. (1990): The Journal of international medical research. 18:201-209.
- Titus F, et al. (1986): Eur Neurol. 25:327-329.
- Sutanto CN, et al. (2024): Clin Nutr. 43:593-602.
- Adamsen D, et al. (2011): Molecular genetics and metabolism. 102:368-373.
- Ramaekers VT, et al. (2001): Molecular genetics and metabolism. 73:179-187.
- FDA (2019): https://www.fda.gov/drugs/drug-safety-and-availability/fda-issues-guidance-compounding-oral-oxitriptan-5-htp-patients-tetrahydrobiopterin-bh4-deficiency
- Walinder J, et al. (1976): Arch Gen Psychiatry. 33:1384-1389.
- Papakostas GI, et al. (2012): Am J Psychiatry. 169:1267-1274.
- Heninger GR, et al. (1983): Arch Gen Psychiatry. 40:1335-1342.
- Lojko D, et al. (2007): J Affect Disord. 103:253-256.
- Das YT, et al. (2004): Toxicol Lett. 150:111-122.
- Byerley WF, et al. (1987): J Clin Psychopharmacol. 7:127-137.
- Bauer M, et al. (2003): Can J Psychiatry. 48:440-448.
- Newman ME, et al. (2000): Int J Neuropsychopharmacol. 3:187-191.
- Mirza NR, et al. (2007): Prog Neuropsychopharmacol Biol Psychiatry. 31:858-866.
- Hansen JY, et al. (2022): Nature neuroscience. 25:1569-1581.
- Haahr ME, et al. (2014): Mol Psychiatry. 19:427-432.
- Nord M, et al. (2013): Int J Neuropsychopharmacol. 16:1577-1586.
- Selvaraj S, et al. (2012): Mol Psychiatry. 17:1254-1260.
- Trivedi MH, et al. (2006): Am J Psychiatry. 163:28-40.
- Soomro GM, et al. (2008): The Cochrane database of systematic reviews. 2008:CD001765.
- Lader M, et al. (2004): Depress Anxiety. 19:241-248.
- Sorensen A, et al. (2022): Mol Psychiatry. 27:192-201.
- Bosker FJ, et al. (2010): Neurochem Int. 57:948-957.
- Goodwin GM, et al. (2023): J Affect Disord. 328:1-5.
- Davis AK, et al. (2020): JAMA psychiatry.
- Ledwos N, et al. (2022): Journal of Clinical Psychopharmacology. 42:581-588.
- Jacobsen JP, et al. (2016): Neuropsychopharmacology. 41:2324-2334.
- Perry KW, et al. (1993): J Pharm Pharmacol. 45:759-761.
- Stahl SM (2008): J Clin Psychiatry. 69:1352-1353.
- Wegener G, et al. (2003): Psychopharmacology. 166:188-194.
- Bauer M, et al. (2021): J Endocrinol Invest. 44:2341-2347.
- Maletic V, et al. (2023): The primary care companion to CNS disorders. 25.
- Jacobsen JPR, et al. (2019): Neuropsychopharmacology. 44:2082-2090.
- Evecxia (2024): Data on file.
- Richard DM, et al. (2009): Int J Tryptophan Res. 2:45-60.
- Papakostas GI, et al. (2014): J Clin Psychiatry. 75:855-863.
- Pan LA, et al. (2017): Am J Psychiatry. 174:42-50.
- Perez-Cruet J, et al. (1971): J Pharmacol Exp Ther. 178:325-330.
- Nagahiro S, et al. (1990): J Cereb Blood Flow Metab. 10:13-21.
- Ercis M, et al. (2023): J Clin Psychiatry. 84.
- Sandrini M, et al. (1996): Life Sci. 58:1551-1559.
- Ito JM, et al. (1977): Neuroendocrinology. 24:55-64.
- Gur E, et al. (1999): J Pharmacol Exp Ther. 288:81-87.
- Singh B, et al. (2022): J Clin Psychiatry. 83.
- Shindo H, et al. (1977): Chem Pharm Bull (Tokyo). 25:1417-1425.
- Hagberg GE, et al. (2002): J Cereb Blood Flow Metab. 22:1352-1366.
- Yang KC, et al. (2018): Transl Psychiatry. 8:132.
- Sternberg EM, et al. (1980): The New England journal of medicine. 303:782-787.
- Joly P, et al. (1991): J Am Acad Dermatol. 25:332-333.
- Hamaguchi Y (2022): Allergol Int. 71:163-168.
- Preuss HG, et al. (2006): Toxicol Mech Methods. 16:281-286.
- Haberzettl R, et al. (2013): Behav Brain Res. 256:328-345.
- Sweet DV (1997).
- Gijsman HJ, et al. (2002): J Clin Psychopharmacol. 22:183-189.
- FDA (2022): https://www.fda.gov/consumers/consumer-updates/fda-101-dietary-supplements
- Cunningham AB, et al. (2021): J Ethnopharmacol. 278:114202.
-